Why does alpha-1 antitrypsin deficiency cause liver disease?

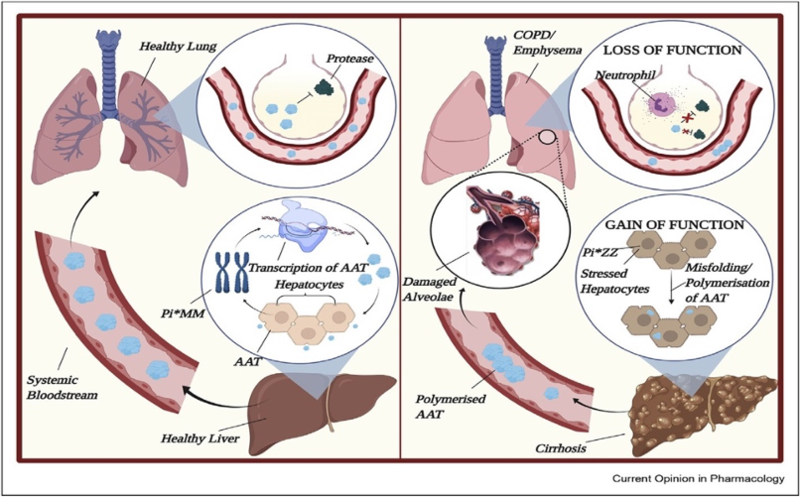
Alpha-1 antitrypsin deficiency (A1AD) is caused by mutations in the SERPINA1 gene, which leads to decreased levels of alpha-1 antitrypsin (A1A), resulting in both lung and liver disease. The pathogenesis of lung and liver disease related to A1AD is markedly different. Lung disease develops when the lack of A1A in the alveoli leads to uninhibited elastase activity causing lung connective tissue breakdown and development of emphysema. However, liver injury results from the intracellular accumulation of misfolded A1A protein within the hepatocyte endoplasmic reticulum (ER), thus inhibiting its secretion. Ultimately, this accumulation can result in liver cell injury and death.
What is alpha-1 antitrypsin?
The serpin family A member 1 gene (SERPINA) encodes the glycoprotein A1A that is synthesized primarily in the hepatocytes and then secreted into the bloodstream. The liver produces approximately 34 mg of A1A per kilogram of body weight per day, leading to a plasma level of 0.9 to 1.75 mg per milliliter, with a half-life of 3 to 5 days. Its function is to protect host tissues by inhibiting neutrophil proteases released non-specifically during inflammation and phagocytosis.
What is the pathogenesis of alpha-1 antitrypsin deficiency?
Inherited mutations in the SERPINA1 gene interfere with A1A production and/or secretion causing a disease termed A1AD.It was first described in 1963, but it was not until the 1970s that an association between A1AD and cirrhosis was recognized. For the last half century, extensive research has been done to understand the pathophysiology and clinical course of lung and liver disease associated with A1AD.


The normal genetic variant for the A1A gene is designated M, and the predominant phenotype in the US population is MM. Almost 95% of severe cases of A1AD are due to a homozygous glutamic acid to lysine substitution at position 342 (Glu342Lys) in the SERPINA1 gene that leads to a misfolded protein. This genetic variant is termed Z and the homozygous phenotype ZZ leads to the most symptoms. The overall prevalence of homozygous individuals is approximately 1:3,000 in the US; over 3 million individuals are affected worldwide.
The heterozygous MZ phenotype prevalence is 2%-4% in Europe and 2%-7.1% in North America. The risk of liver disease in these persons is possibly increased by the presence of additional coexisting factors like alcohol abuse, nonalcoholic fatty liver disease (NAFLD) and cystic fibrosis.
Other alleles have been described that range from having no effect on the level of functioning to lack of protein function. Rare alleles, MMalton and MDuarte have also been associated with liver disease, however the S and null variants are not associated with liver disease.

What is the natural history of alpha-1 antitrypsin deficiency?
The natural history of A1AD is variable and not completely understood. There is considerable variability in phenotypic expression in both lung and liver disease. The risk of developing lung and liver disease depends not only on which deficient alleles the individual carries, but also genetic polymorphisms and environmental factors.
In most patients the dominant manifestation is lung disease, which begins in early adulthood. Noticeably, adults with severe lung disease often do not develop liver disease and vice-versa. However, it has been shown that in adults, hepatic disease can coexist with pulmonary emphysema. In a study that included 57 patients with the ZZ phenotype and established pulmonary disease, 63.2% had findings suggestive of liver disease and 17.5% showed evidence of advanced liver fibrosis.
A1AD has been associated with a spectrum of liver-related pathologies, ranging from neonatal hepatitis and jaundice to chronic hepatitis, to cirrhosis and end-stage liver disease including hepatocellular carcinoma (HCC). While as many as 73% of infants with A1AD have transient elevation of liver enzymes, only 11% and 6% of infants develop prolonged cholestatic jaundice and chronic liver disease respectively. In adults, chronic liver disease progresses to cirrhosis in 10% of patients with A1AD, 1.3% develop HCC and 14.7% require liver transplantation.
Recent studies have found significant genomic hypomethylation in A1AD liver-impacting genes related to liver cancer, cell cycle, and fibrosis, as well as key regulatory molecules influencing growth, migration, and immune function. These factors could account for some of the heterogeneity in clinical expression of A1AD. Other environmental and genetic factors may influence the frequency and rate of progression of liver disease.

What is our understanding of the liver injury mechanism in A1AD?
Following, is a proposed chronology of events leading to liver injury associated with A1AD:
- Transcription and translation of the A1A mutant Z gene by the normal pathways
- Translocation of the polypeptide chain into the ER. However, the protein molecule does not fold properly into its final conformation.
- Chaperone proteins in the ER recognize these mutant Z molecules as abnormal and direct them to a series of proteolytic pathways rather than allowing their secretion.
- It is thought that the proteasomal pathways [as a part of ER-associated degradation (ERAD)] are the primary route for degradation of A1A mutant Z in the non-polymerized conformation.
- For unclear reasons, some of the mutant Z molecules escape proteolysis and may adopt a polymer conformation that is highly thermodynamically stable and links large groups of mutant Z molecules together with non-covalent bonds.
- Autophagy then becomes the primary route of degradation. This is a highly conserved degradation system in which specialized vacuoles degrade abnormal proteins and larger structures, such as senescent organelles. The mutant Z protein is directed into autophagic vacuoles. A1A mutant Z protein polymers have increased accumulation in autophagy-deficient systems.
- The reduced efficiency of degradation in liver disease patients is thought to be due to a greater steady state burden of mutant Z protein within liver cells and increased liver injury.
Liver injury is a slow process that takes place over years, with analysis of human livers showing that accumulation of A1A mutant Z protein is very heterogeneous among individual hepatocytes. ERAD and autography processes become overwhelmed and then A1A polymerization and accumulation occurs. Hepatocytes with the largest burden of A1A mutant Z are most susceptible to apoptosis.
Polymer accumulation correlates with fibrosis stage. This cycle of stress, death and repair ultimately can lead to liver fibrosis, cirrhosis and hepatocellular carcinoma. Environmental and genetic modifiers of protein secretion, degradation, apoptosis or regeneration would then be hypothesized to influence the progression of liver disease in an individual patient.

Risk factors for liver disease in patients with the ZZ phenotype include male sex, age >50, repeatedly elevated liver enzymes, obesity, diabetes and metabolic syndrome. Reported prevalence of cirrhosis is highly variable (2-43%) depending on population studied. Recent cirrhosis estimates 7-10% with very limited survival estimated at median of 1.8 years. 1-year survival probability is 55%.
What is the risk of liver disease associated with MZ phenotype?
In total, approximately 120 million people worldwide carry at least one Z allele. In recent years, there has been an enormous interest to determine if the MZ phenotype precipitates the development of liver disease, but this remains controversial.
New research showed that patients with the MZ phenotype have higher levels of serum transaminases, A1A inclusions in the liver and liver stiffness compared to patients without the mutation. Also, it was found that patients with cirrhosis and the MZ phenotype developed complications from cirrhosis faster.
Nonetheless, the absolute risk of liver disease among adults with MZ phenotype is unknown, but the risk is increased by the presence of additional coexisting conditions, such as NAFLD, alcohol misuse, and cystic fibrosis. A recent study showed that the MZ phenotype confers an approximately six times higher odd to develop both NAFLD and alcohol-associated cirrhosis. However, further studies are necessary to characterize the direct contribution of the phenotype in the development of cirrhosis.
How do we diagnose alpha-1 antitrypsin deficiency?
The gold standard for the diagnosis of A1AD is the genotype analysis of genomic DNA in a serum or buccal mucosa sample or the analysis of the phenotype protein in the patient’s serum. Some providers use the serum A1A level as screening test and then perform the gold standard test if levels are abnormal.
Liver biopsy is not required to establish the diagnosis of A1AD but can be useful to understand the degree of liver injury. The signature feature of A1AD on a liver biopsy specimen is the presence of periodic acid-Schiff-positive (PAS1), diastase-resistant globules. The globules represent the polymerization of the abnormally folded Z protein retained within the hepatocyte.

How do we screen for A1AD-associated liver disease?
Guidelines for screening for liver disease recommend to screen with liver ultrasound and laboratory monitoring of aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyltransferase (GGT), albumin, bilirubin, international normalized ratio (INR), and platelets. Unfortunately, adherence to guidelines is poor with only 45% of patients with A1A ZZ having annual liver enzyme evaluation and 21% regular ultrasound of the liver.New research found that the sensitivity of an ALT above the upper limit of normal was poor for the detection of liver disease and suggesting that monitor of serum transaminases is not helpful to screen for liver disease.
How do we treat A1AD-associated liver disease?
Exogenous A1A replacement, which is commonly used to treat A1AD-associated emphysema, has no effect on the development of liver disease. Liver transplantation remains the only curative treatment option but only considered when liver failure develops. The clinical spectrum of patients with A1AD undergoing liver transplantation is wide, from young children to adults in their fifties. Long-term outcomes after liver transplantation or combined lung-liver transplantation for A1AD are favorable and like those for other indications.
One issue that has not been extensively studied, is the evolution of chronic obstructive pulmonary disease after liver transplantation. Despite the normalization of A1A levels after liver transplantation, the forced expiratory volume in 1 second (FEV1) continues to decline for most patients with A1A mutant protein Z. Individual data suggest a continued decline in 65% with an improvement in 35% with wide variations. More importantly, 53% had an annual reduction in FEV1 greater than the expected 30 mL/y from aging. This obviously has implications for patient long-term morbidity and mortality after liver transplant.
Currently there are no approved pharmacological treatment for A1AD-related liver disease, but there are several promising approaches that are undergoing clinical testing. The therapeutic approaches aim to reduce the intrahepatic Z-A1A burden by one of the following strategies:
- Degrade: Augment autography to clear inclusions
- Small molecule: Block intracellular polymerization
- Small interfering RNA (siRNA): Silence expression of Z
- Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR): Correct defect with gene therapy
Carbamazepine, a drug known to be well tolerated in humans, has shown to clear A1A inclusions by increasing degradation in a mouse model of A1AD which provided a proof for therapeutic use of autography enhancers.
siRNA can regulate gene expression and has the potential to silence specific genes in targeted cell types. Fazirsiran is a RNAi therapeutic that contains asynthetic, double-stranded, small interfering RNA duplex conjugated to N-acetylgalactosamine, which binds to the hepatocyte asialoglycoprotein receptor to facilitate endosomal uptake and intracellular delivery. In a recent phase 2 open label small trial, Fazirsiran reduced the production and accumulation of intrahepatic A1A mutant protein Z associated with a strong reduction of its concentration in the serum and liver and improvements in liver enzymes. Larger and longer clinical trials are needed to determine the effect of fazirsiran on fibrosis.

Key Points
- A1AD is a toxic gain of function mutation in the liver and loss of function mutation in the lungs.
- The retention of the misfolded protein within the endoplasmic reticulum of the hepatocyte is the inciting event in the pathogenesis of liver disease. Endoplasmic reticulum–associated degradation and autophagy degrade the misfolded protein, but when the capacity of these processes is overwhelmed, polymerization and accumulation occur. This ultimately leads to cell death, liver fibrosis, and cirrhosis.
- New therapies are on the horizon.