“Glycogenosis” on Liver Biopsy: Clinicopathologic Considerations

Case #1: A 23-year-old patient presents with hepatomegaly and transaminitis. Liver function tests include alkaline phosphatase 164 U/L, AST 77 U/L, ALT 63 U/L, and total bilirubin 0.2 mg/dL. History is notable for poorly controlled type 1 diabetes in the setting of numerous psychosocial stressors and barriers to care; HbA1c is 12.9% and the patient has had multiple hospital admissions for diabetic ketoacidosis in the past 5 years. Ceruloplasmin is within normal limits. BMI is 25.3 kg/m2.The patient undergoes liver biopsy.
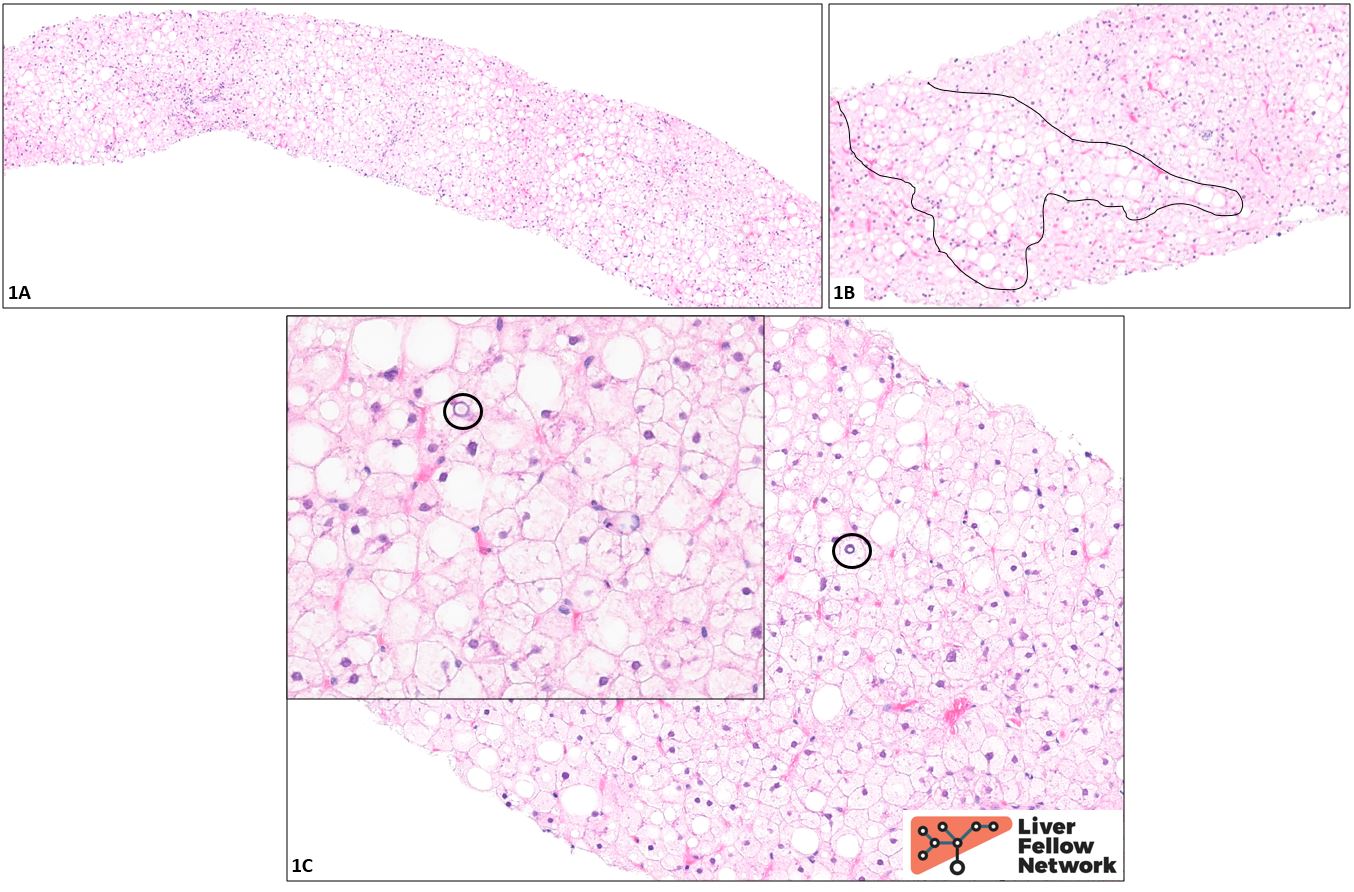
Histologic findings and diagnosis: At low power (1A), the biopsy appears rather “pale.” Higher power examination (1B) reveals ~40% macrovesicular steatosis (representative area circled in black). The remaining hepatocytes appear diffusely swollen and show a striking degree of cytoplasmic rarefication and clearing. The nuclei are generally small and dark (“pyknotic”), though a few are somewhat enlarged and contain pale inclusions (black circles, 1C and inset), imparting a “cleared-out” appearance. No fibrosis is seen on the trichrome and reticulin stains (not shown).
Although the distinctive pale, enlarged cells that comprise most of the parenchyma are reminiscent of the balloon cells of steatohepatitis, it should be noted that balloon cells are generally scattered throughout a biopsy, rather than present in the high number and density seen here. In addition, no Mallory-Denk bodies (eosinophilic clumps of degraded cytoskeletal material sometimes seen in the cytoplasm of balloon cells) are identified, though this is not a particularly sensitive criterion. Overall, therefore, these cells do not appear to be balloon cells: this is not a case of steatohepatitis. In a young patient with poorly controlled type 1 diabetes, the cells are instead likely to be distended with glycogen, and indeed, the findings here are classic for glycogenic hepatopathy.
Case #2: A 60-year-old patient undergoes orthotopic liver transplantation in the setting of decompensated cirrhosis. History is further notable for longstanding diffuse muscle weakness. Liver function tests at the time of transplantation are as follows: alkaline phosphatase 203 U/L, AST 81 U/L, ALT 39 U/L, and total bilirubin 0.6 mg/dL.
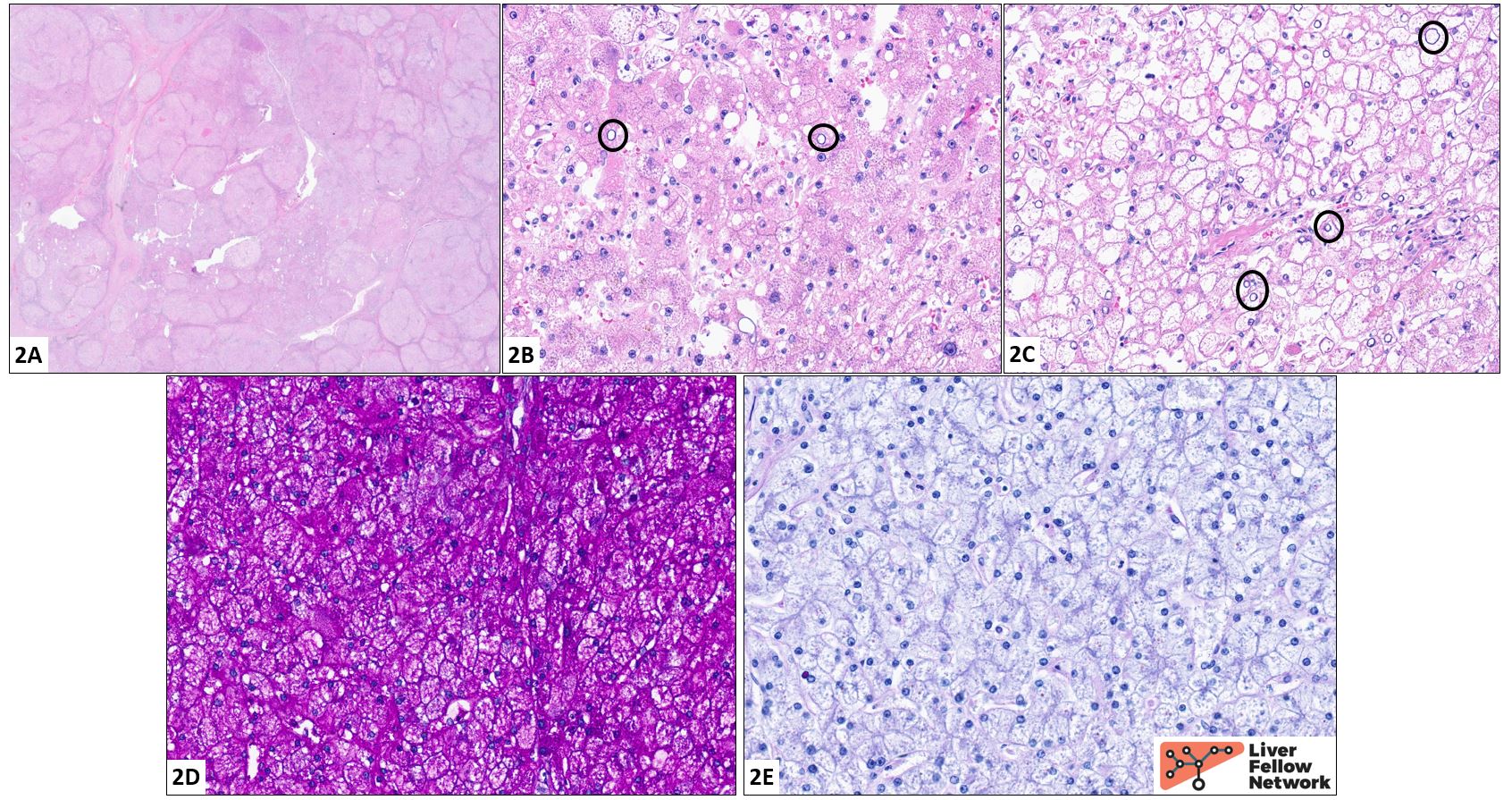
Histologic findings and diagnosis: Sections of the explanted liver reveal a mixed macro/micronodular pattern of cirrhosis (2A). The hepatocytes (2B, 2C) are diffusely enlarged and frequently exhibit cytoplasmic engorgement with a pale eosinophilic granular material. The cell borders are crisp, resulting in a "cobblestone" appearance (best seen in 2C). Nuclear clearing is readily identified (black circles). The cytoplasmic contents stain a vivid magenta color with a periodic acid Schiff stain (PAS stain, 2D); this is lost following digestion with diastase (PAS/D stain, 2E). These findings, in conjunction with the clinical history, fit well for a diagnosis of glycogen storage disease.
Genetic workup revealed a homozygous c.4322_4323dupAA pathogenic variant in the AGL gene, consistent with type III glycogen storage disease. The history of muscle weakness is suggestive of subtype IIIa.
Overview
These opening vignettes highlight the liver’s role as a “glucostat” – an essential regulator of carbohydrate metabolism, including storage of carbohydrates in the form of glycogen (glycogen synthesis or glycogenesis), breakdown of glycogen and release of constituent glucose into the bloodstream (glycogenolysis), and de novo synthesis of glucose (gluconeogenesis). At baseline, normal hepatocytes contain a modest amount of glycogen, stored primarily in the cytoplasm (90%) and, to a lesser extent, within the lysosomes.
Normal hepatocellular glycogen stores are insufficient to be appreciated on H&E but do stain a vivid magenta or fuchsia color with PAS (Figure 2A). Addition of diastase (alpha-amylase) to a PAS stain converts glycogen into water-soluble maltose, resulting in loss of staining (Figure 2B). In contrast, hepatocytes containing excess glycogen may show a spectrum of changes appreciable even on H&E, though it should be noted that these are quite nonspecific. These features, collectively known as glycogenosis, are illustrated in the cases above and include:
- Diffuse hepatocellular distension due to engorgement by glycogen; cell membranes are often crisp and well-delineated, resulting in a “cobblestone” appearance
- Hepatocyte swelling may result in sinusoidal compression
- Pale or “rarefied” cytoplasm
- Glycogenated nuclei (glycogen-rich nuclear vacuoles), giving the nuclei a “hollowed-out” appearance
The clinical differential for excessive hepatic glycogen on liver biopsy includes glycogenic hepatopathy (essentially, glycogenosis and hepatomegaly in the setting of severe hyperglycemia) versus the various glycogen storage disorders (GSDs).1,3–5 Urea cycle disorders (UCDs) may also be considered, though the histologic findings appear highly variable based on the limited available data. Differentiating among these entities is virtually impossible on the basis of histology alone: clinical correlation is essential. In the case of heritable disorders, genetic testing is often crucial for confirmation of the diagnosis and disease subtyping. In this post, we briefly review the clinicopathologic features of these entities.
Glycogenic hepatopathy
Glycogenic hepatopathy, knownvariouslythrough the years as diabetes mellitus-associated glycogen storage, [diabetes-associated] hepatomegaly, and hepatic glycogenosis, occurs in the setting of severe hyperglycemia alternating with relative, and typically exogenous, hyperinsulinemia. The pathophysiology is thought to involve diffusion of glucose into hepatocytes during protracted periods of hyperglycemia, followed by sporadic glycogen synthesis during insulin spikes.
Patients present with transaminitis of fluctuating severity and potentially massive hepatomegaly; ascites has been reported, possibly due to sinusoidal compression by the engorged hepatocytes. Of note, ultrasound findings in glycogenic hepatopathy may overlap with those of fatty liver disease, as both appear echogenic; biopsy is necessary to distinguish between the two. Biopsies display the classic features of excess hepatocellular glycogen, such as distended hepatocytes with glycogenated nuclei. Steatosis/steatohepatitis is uncommon (<20%). Crucially, and in contrast to fatty liver disease, progression to fibrosis is not seen in glycogenic hepatopathy: indeed, the presence of fibrosis in a liver with significant glycogen accumulation argues for a hereditary glycogen storage disorder and against glycogenic hepatopathy.
The prototypical clinical correlate of glycogenic hepatopathy is, of course, poorly controlled type 1 diabetes mellitus in a young patient with a history of DKA, as seen in vignette #1. In extreme cases, patients may present with Mauriac syndrome, a constellation of glycogenic hepatopathy, delays in growth and puberty, hypercholesterolemia, and a cushingoid appearance. Glycogenic hepatopathy has more recently been described in a number of other clinical settings, such as type 2 diabetes mellitus requiring insulin therapy, high-dose steroid bursts, and dumping syndrome. Interestingly, similar findings may be seen in rare cases of anorexia nervosa, though the mechanism is unclear. Liver function tests and biopsy findings appear to resolve with improved glycemic control.
Glycogen storage diseases
The glycogen storage diseases (GSDs) or glycogenoses are monogenic disorders characterized by deficiencies in the enzymes governing the glycogen synthesis, glycolysis, or glycogenolysis pathways. Much of the body’s glycogen is stored in the liver and in skeletal muscle, and thus these are the organ systems most commonly affected in GSDs, though other organs (heart, brain, kidney) may also be implicated.
Liver biopsy findings vary based on the underlying GSD, but recurring themes include “glycogenosis” (i.e., pale cytoplasm and glycogenated nuclei, as described above) and steatosis; certain GSDs carry a risk of fibrosis/cirrhosis. Given the nonspecific nature of these findings, clinical correlation (often including genetic testing) is essential. Clinically, the common denominator is fasting hypoglycemia and hepatomegaly (with the notable exception of type 0); muscle involvement may manifest as exercise intolerance, muscle cramps or weakness, and rhabdomyolysis. Age at presentation is highly variable and may range from infancy to adulthood. The table below lists the clinicopathologic findings associated with several of the more common so-called “hepatic GSDs.”
Clinical management varies based on the underlying disorder and is beyond the scope of this post; broadly speaking, many GSDs are managed via high-protein meals during the day and uncooked cornstarch prior to periods of fasting (e.g., during sleep). In some instances (GSD-VI, IX), the clinical course is fairly benign, and symptoms tend to regress as patients reach adulthood. Enzyme replacement therapy (alglucosidase alfa) has been introduced for GSD-II (Pompe disease). Liver transplantation may be necessary in cases of advanced fibrosis, e.g., in the rapidly progressing form of GSD-IV.
Urea cycle defects
Urea cycle defects (UCDs) classically encompass deficiencies in arginase (ARG1), argininosuccinate acid lyase (ASL), argininosuccinic acid synthetase (ASS), carbamoyl phosphate synthetase I (CPS 1), N-acetylglutamate synthetase (NAGS), and ornithine transcarbamylase (OTC). Liver histopathology in these rare disorders varies widely; the few available case series describe steatosis, fibrosis and, in some instances, focal or diffuse glycogenosis. Clear-cut genotype/phenotype associations have not been delineated; to date, evidence of excessive hepatocellular glycogen has been reported, at least focally, in several of the UCDs, including ASL and OTC deficiencies. As such, though rare, UCD should be considered in the differential for excessive hepatic glycogen in young patients who present with neurologic symptoms, metabolic derangement (particularly hyperammonemia), and/or protein aversion.
Final thoughts
As seen here, excessive glycogen in a liver biopsy is a nonspecific finding; in addition, only some of the “classic” histologic features may be appreciated on biopsy, while others (such as glycogenated nuclei) may be only focal and thus more difficult to identify. Close correlation with clinical history is key.
Clinicopathologic Features of Glycogen Storage Disorders with Hepatic Involvement
Table adapted from Torbenson’s Biopsy Interpretation of the Liver and Atlas of Liver Pathology and from Gümüş and Özen (2023); see also Özen (2007) and Ellingwood and Cheng (2018)
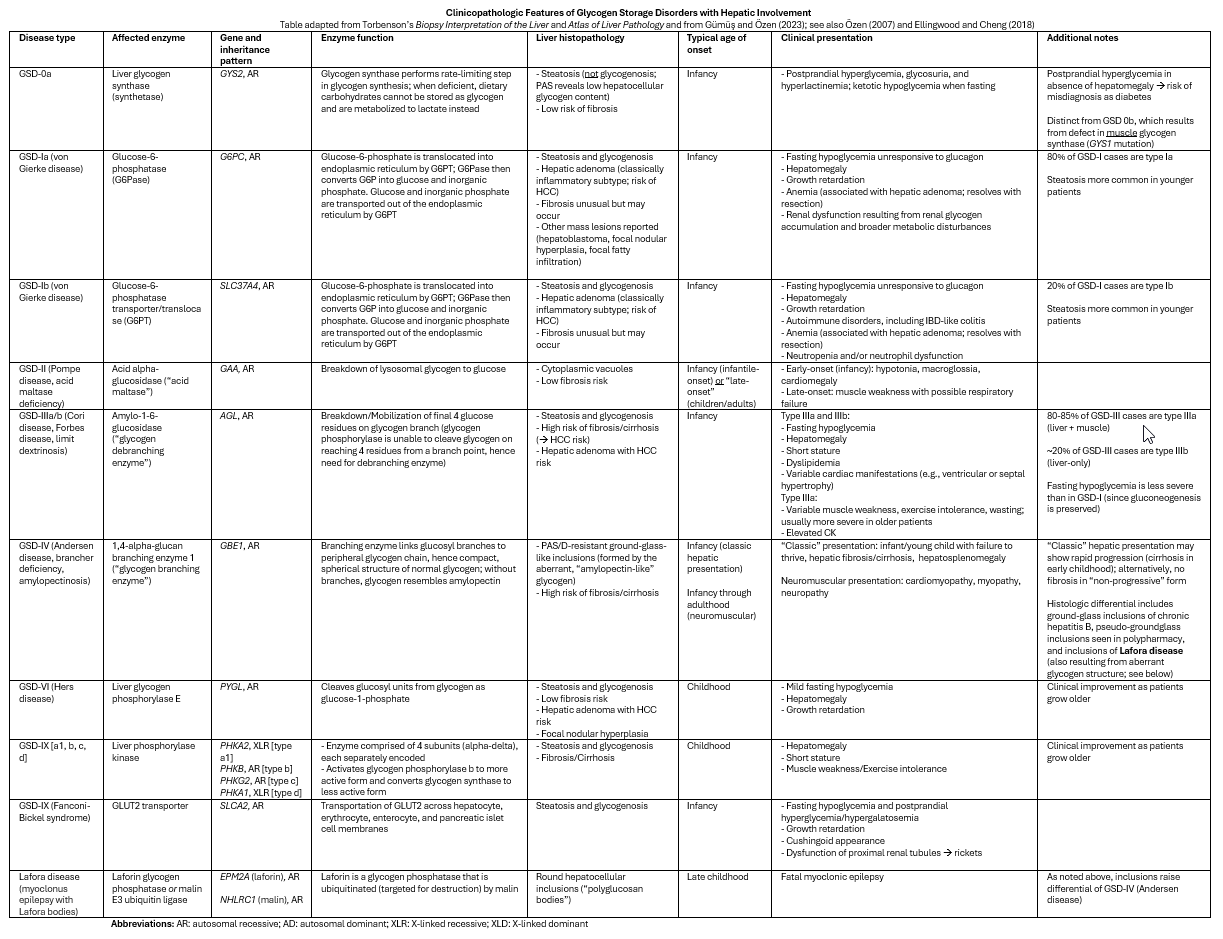
Abbreviations: AR: autosomal recessive; AD: autosomal dominant; XLR: X-linked recessive; XLD: X-linked dominant
References
Badizadegan K, Perez-Atayde AR. Focal glycogenosis of the liver in disorders of ureagenesis: its occurrence and diagnostic significance. Hepatol Baltim. 1997;26(2):365-373. doi:10.1002/hep.510260217
Bigot A, Tchan MC, Thoreau B, et al. Liver involvement in urea cycle disorders: a review of the literature. J Inherit Metab Dis. 2017;40(6):757-769. doi:10.1007/s10545-017-0088-5
Bronstein HD, Kantrowitz PA, Schaffner F. Marked enlargement of the liver and transient ascites associated with the treatment of diabetic acidosis. N Engl J Med. 1959;261:1314-1318. doi:10.1056/NEJM195912242612603
Chatila R, West AB. Hepatomegaly and abnormal liver tests due to glycogenosis in adults with diabetes. Medicine (Baltimore). 1996;75(6):327-333. doi:10.1097/00005792-199611000-00003
Cox BK, Guindi M, Hutchings D, et al. Glycogenic hepatopathy is associated with type 1 diabetes mellitus in only a minority of cases in a contemporary adult population. Ann Diagn Pathol. 2023;64:152130. doi:10.1016/j.anndiagpath.2023.152130
Ellingwood SS, Cheng A. Biochemical and clinical aspects of glycogen storage diseases. J Endocrinol. 2018;238(3):R131-R141. doi:10.1530/JOE-18-0120
Fitzpatrick E, Cotoi C, Quaglia A, Sakellariou S, et al. Hepatopathy of Mauriac syndrome: a retrospective review from a tertiary liver centre. Arch Dis Child. 2014;99(4):354-357. doi:10.1136/archdischild-2013-304426
Gümüş E, Özen H. Glycogen storage diseases: an update. World J Gastroenterol. 2023;29(25):3932-3963. doi:10.3748/wjg.v29.i25.3932
Hannah WB, Derks TGJ, Drumm ML, et al. Glycogen storage diseases. Nat Rev Dis Primer. 2023;9(1):46. doi:10.1038/s41572-023-00456-z
Iancu TC, Shiloh H, Dembo L. Hepatomegaly following short-term high-dose steroid therapy. J Pediatr Gastroenterol Nutr. 1986;5(1):41-46. doi:10.1097/00005176-198601000-00008
Komatsu M, Yazaki M, Tanaka N, et al. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol. 2008;49(5):810-820. doi:10.1016/j.jhep.2008.05.016
Komuta M, Harada M, Ueno T, et al. Unusual accumulation of glycogen in liver parenchymal cells in a patient with anorexia nervosa. Intern Med Tokyo Jpn. 1998;37(8):678-682. doi:10.2169/internalmedicine.37.678
Miles L, Heubi JE, Bove KE. Hepatocyte glycogen accumulation in patients undergoing dietary management of urea cycle defects mimics storage disease. J Pediatr Gastroenterol Nutr. 2005;40(4):471-476. doi:10.1097/01.mpg.0000157200.33486.ce
Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25(1):21-29. doi:10.1016/s0887-8994(00)00276-9
Nishimura R, Ishak K, Reddick R, et al. Lafora’s disease: diagnosis by liver biopsy. Trans Am Neurol Assoc. 1979;104:119-120.
Ozen H. Glycogen storage diseases: new perspectives. World J Gastroenterol. 2007;13(18):2541-2553. doi:10.3748/wjg.v13.i18.2541
Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572-587. doi:10.1038/nrendo.2017.80
Pinto MJF, Melo N, Flores L, et al. Mauriac syndrome: a rare complication of type 1 diabetes mellitus. Eur J Case Rep Intern Med. 2018;5(12):000969. doi:10.12890/2018_000969
Ranucci G, Rigoldi M, Cotugno G, et al. Chronic liver involvement in urea cycle disorders. J Inherit Metab Dis. 2019;42(6):1118-1127. doi:10.1002/jimd.12144
Resnick JM, Zador I, Fish DL. Dumping syndrome, a cause of acquired glycogenic hepatopathy. Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc. 2011;14(4):318-321. doi:10.2350/10-07-0876-CR.1
Sherigar JM, Castro JD, Yin YM, et al. Glycogenic hepatopathy: a narrative review. World J Hepatol. 2018;10(2):172-185. doi:10.4254/wjh.v10.i2.172
Soon GST, Torbenson M. The liver and glycogen: in sickness and in health. Int J Mol Sci. 2023;24(7):6133. doi:10.3390/ijms24076133
Torbenson M, Chen YY, Brunt E, et al. Glycogenic hepatopathy: an underrecognized hepatic complication of diabetes mellitus. Am J Surg Pathol. 2006;30(4):508.
Torbenson M. Glycogenic Abnormalities on Liver Biopsy. In: Ferrell LD, Karkar S, eds. Liver Pathology. Consultant Pathology. New York: Demos Medical Publishing; 2011.
Torbenson MS. Atlas of Liver Pathology: a Pattern-Based Approach. Philadelphia: Wolters Kluwer; 2020.
Torbenson MS. Biopsy Interpretation of the Liver. 4th ed. Philadelphia: Wolters Kluwer; 2021.
van den Brand M, Elving LD, et al. Glycogenic hepatopathy: a rare cause of elevated serum transaminases in diabetes mellitus. Neth J Med. 2009;67(11):394-396.
Verhalen B, Arnold S, Minassian BA. Lafora disease: a review of molecular mechanisms and pathology. Neuropediatrics. 2018;49(6):357-362. doi:10.1055/s-0038-1675238
Yaplito-Lee J, Chow CW, Boneh A. Histopathological findings in livers of patients with urea cycle disorders. Mol Genet Metab. 2013;108(3):161-165. doi:10.1016/j.ymgme.2013.01.006